Solid Solutions and Dispersions
Two significant challenges in drug delivery are, one, enhancing the bioavailability of poorly water-soluble active pharmaceutical ingredients (APIs) and, two, controlling the release of APIs from drug-eluting devices. Solid solutions and dispersions are used in many routes of drug administration, such as oral, mucosal (vaginal, rectal, buccal, ocular), subcutaneous, subdermal, and transdermal, and can help alleviate some of these issues.
Methods of Preparation
Solid solutions and dispersions are generally made by the “melt method” or the “solution method”. The excipients in these formulations are usually polymers, but small molecules such as urea¸ sucrose, dextrose, and galactose have been used, as have waxes.
In the solution method, API and excipients are co-dissolved in a solvent, which is then removed to form the dosage form. This method is used to form slabs of material which can be ground into powders. It is also the basis of spray drying and emulsification/solvent removal. Spray drying involves forcing an aqueous or organic solution of API and excipients through a nozzle to form an aerosol in an evaporation chamber. The solvent evaporates, leaving solid API/excipient particles. The API in the particles can be crystalline, amorphous, or can show characteristics of both1. Emulsification/solvent removal involves preparing a solution of API and excipients in a water-immiscible solvent, which is then emulsified into an aqueous surfactant solution. The solvent is removed from the emulsion droplets to form particles. Water insoluble polymers are most commonly used in this process, for example, PLA, PLGA, ethyl cellulose, or cellulose acetate phthalate.
In the melt method, API is added to molten excipients and the mixture is then cooled. This method is the basis of spray chilling (spray congealing), melt/ emulsification/chill-hardening, hot-melt extrusion, and injection molding. Spray chilling is analogous to spray drying, but the API/excipient mixture is molten rather than in solution and the final particles are formed by solidification of the cooling aerosol droplets rather than by solvent evaporation. Waxy excipients such as Compritol®, Dynasan®, and lecithin are especially useful in this method. Melt/emulsification/chill hardening is analogous to emulsification/solvent removal except that the final particles are formed by cooling of the emulsified melt.
Hot-melt extrusion is becoming widely practiced in the pharmaceutical industry. Thermoplastic polymers such as polyethylene glycol, polyvinylpyrrolidinone, various lactic acid-glycolic acid copolymers, various acrylates, and poly(vinylcaprolactam-covinyl acetate-g-polyethylene glycol) are co-melted with the API in a hot-melt extruder and the extrudate is further processed into the final dosage form2.
Injection molding is used in the manufacture of some drug-eluting devices and has also been used to prepare solid oral dosage forms3,4. The API is mixed with thermoplastic excipients and injected molten into a mold where the final dosage form is made upon cooling.
Solid Solution and Dispersion Types
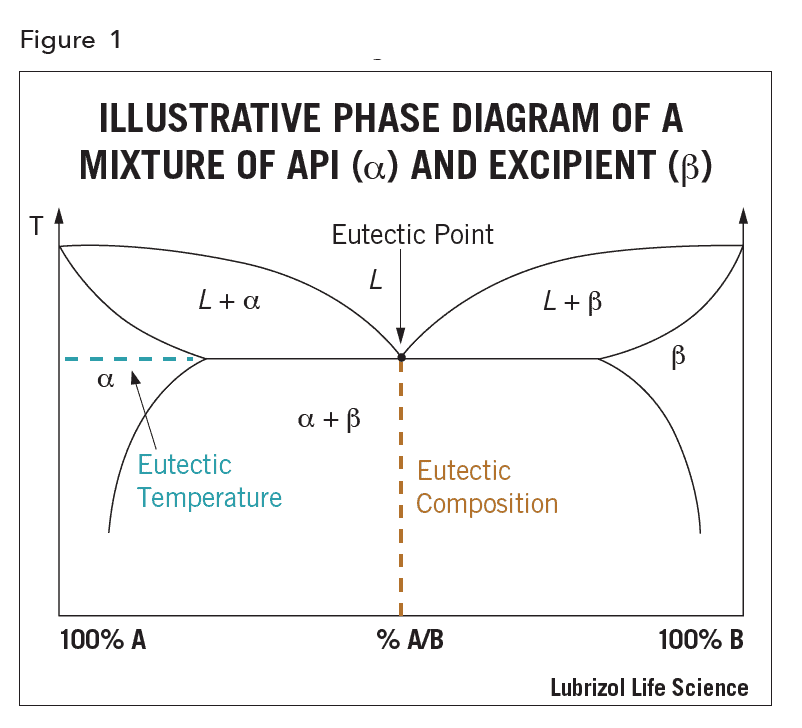
The drug may be molecularly dissolved in the solid excipient matrix (i.e. solid solution) or dispersed as crystalline or amorphous particles (i.e. solid dispersion). If dissolved, the API could in principle be miscible with the excipients over the whole composition range, however, in practice, the API and excipients are miscible over only a limited range, as illustrated by the phase diagram in Figure 1. The eutectic composition has the lowest melting point, called the eutectic temperature, and the eutectic point is the intersection of the eutectic composition and eutectic temperature in the phase diagram.
In crystalline solid solutions the API can occupy crystal lattice sites or the interstitial spaces. If the formulation is amorphous, such as when the API is dispersed in an amorphous polymer, the API is distributed at random between the excipient molecules and can be present as crystalline or amorphous particles, or in molecular solution.
 Drug Release Mechanisms and Bioavailability
Drug Release Mechanisms and Bioavailability
The mechanisms of API release from solid solutions and dispersions include: (1) dissolution of the entire dosage form, if the excipients are soluble, (2) dissolution of API from within channels in insoluble excipients, leaving the excipients more-or-less intact, and (3) molecular diffusion of the API through the bulk of the dosage form, also leaving the dosage form physically intact.
The bioavailability of an API largely depends on how it is incorporated into the drug product, the type of solid dosage form (solution or dispersion), and the mechanism of release. For example, since absorption of API requires the API to be in solution in the body, the oral bioavailability of a poorly water-soluble API can be improved by increasing its rate of dissolution according to the Noyes-Whitney equation:
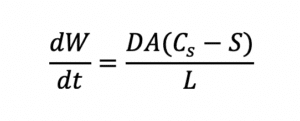
Since dissolution rate is proportional to API surface area (A), the larger the total surface area of a given dose of API, the higher the rate of dissolution and bioavailability for a solid solution. Bioavailability is also related to API solubility. Smaller particles not only dissolve faster than larger particles, but they are also more soluble than their larger counterparts, as shown by the Ostwald-Freundlich equation:
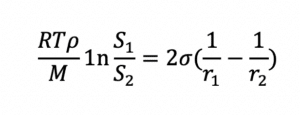
Water-soluble polymers and surfactants used to form the solid solution or dispersion increase the water solubility of the API, improving its bioavailability further.
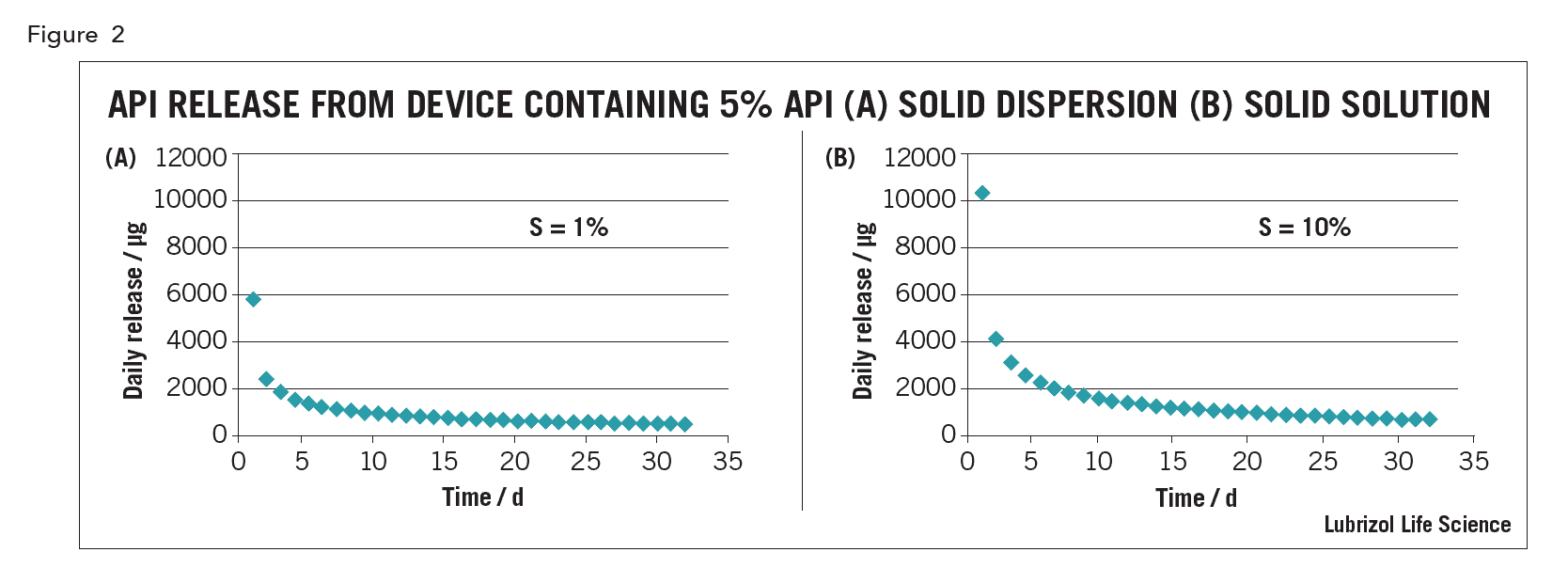
For drug-eluting polymeric implants, whether the drug is in solid solution or solid dispersion affects release kinetics. For example, an intravaginal ring (IVR) containing 5% API, with an API solubility of 1% in the matrix polymer (a solid dispersion), the in vitro release curve under sink conditions predicted using the equations of Helbling is shows in Figure 2(a)5. For a device of the same size with the same 5% loading but made as a solid solution from excipients in which the API solubility is 10% (assuming the same API diffusion coefficient in the device), the drug release predicted using the model of Siepman and Siepman is shown in Figure 2(b) 6. Although both devices have a similarly shaped kinetic curve, drug release from the solution device is higher, especially on day 1, all other things being equal.
 Characterization
Characterization
The most commonly used methods for characterizing solid dispersions are visual, hot stage optical microscopy (OM), differential scanning calorimetry (DSC), and by x-ray diffraction (XRD). The aim of characterization is to determine if the API is in solution or dispersion. If in dispersion, then it is to determine whether the API is in crystalline or amorphous form, whether in one or more polymorphic forms, and to determine the API domain size as this can affect dissolution or release kinetics and hence bioavailability, and also affect long term product stability.
If the solid is transparent, then the API is usually in solution. However, supersaturated forms produced by rapid cooling of viscous precursors that limit the API molecular diffusion and hence crystallization, can also appear transparent, even though the API is above the solubility concentration. Opaque solids are usually API dispersions and are always so when the solid excipients are otherwise transparent.
DSC is used to determine the melting point of an API in the solid and the melting point of the excipient matrix. It can also be used to determine the solubility of an API in the excipients, as the magnitude of the area under the API melting peak on a DSC graph is proportional to the amount of crystalline API in the formulation. Extrapolation of the area to loading curve to zero area can yield the solubility of the API. Care must be taken in interpreting the results of this measurement however, since it is made at the melting temperature of the API, where the solubilizing power of the excipients may be substantial. If the excipients melt and dissolve the API at a temperature below the API melting temperature, no API melt peak will be detected even for high API loadings. This was observed for dapivirine dispersed in ethylene vinyl acetate copolymer up to 40% API, which is well above the equilibrium API solubility of ~0.25% determined from diffusion experiments7. For crystalline excipient matrices and amorphous matrices that melt above the API melting point, this method should be suitable to determine API solubility and will also indicate the presence of polymorphs that have different melting points.
X-ray diffraction yields characteristic peaks for any crystalline components of a formulation. Thus, it can be determined if the API is crystalline or amorphous and can clearly indicate any crystalline polymorphs present. The peak width can also be used to determine the crystal size of the API in the formulation using the Scherrer equation8:
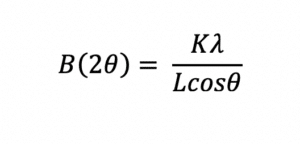
Stability
True solid solutions are thermodynamically stable. Other solid solutions and dispersions can suffer from various instabilities. For example, API initially dissolved in supersaturated solid solutions can precipitate over time. There is no thermodynamic driving force for resolubilization of API that diffuses to the surface of a supersaturated solid solution and subsequently crystallizes. As discussed previously, small particles are more soluble than large particles, so the size distribution of API particles in solid dispersions can evolve over time as large particles grow at the expense of the smaller ones (Ostwald ripening). An API dispersed in amorphous particles are also thermodynamically unstable and can crystallize over time.
References
- Robert Lee, Amir Zalcenstein, and Mark Mitchnick, Solumer™ Technology: a Viable Oral Dosage Form Option for BCS Class II Molecules, ONdrugDelivery, May 2011,
- Soluplus™, BASF
- Quinten et al. Development and evaluation of injectionmolded sustained-release tablets containing ethylcellulose and polyethylene oxide, Drug Development and Industrial Pharmacy, 2011, 37(2):149–159.
- Hemmingsen H., Haahr A.M., Gunnergaard C., Cardot J.-M. Development of a New Type of Prolonged Release Hydrocodone Formulation Based on Egalet®ADPREM Technology Using In Vivo-In Vitro Correlation. Pharmaceutics. 2011; 3(1): 73-87.
- Helbling, M., Luna, J.A., Cabrera, M.I., Mathematical modeling of drug delivery from torusshaped single-layer devices. J. Control. Release 149, 258–263 (2011).
- J. Siepmann, F. Siepmann Modeling of diffusion controlled drug delivery, Journal of Controlled Release (2011) in press.
- Particle Sciences internal communication.
- Scherrer, “Bestimmung der Grösse und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen,” Nachr. Ges. Wiss. Göttingen 26 (1918) 98-100.