Sampling of Powders
Knowledge of the particle size distribution (PSD) of powder systems is a prerequisite for most manufacturing operations, including those associated with the production of pharmaceutical and biopharmaceutical products. If the PSD is not controlled in a process, high rejection rates can result and significant production losses can be incurred. Particle size analytical results are most applicable when samples drawn are representative and the appropriate dispersion tehniques are used. The majority of variation in particle sizing measurements is traceable to either incorrect sampling or sample preparation.
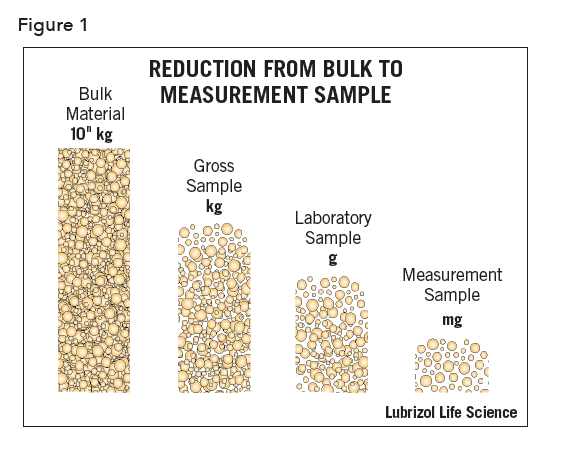
When measuring the PSD of a powdered solid, the results will have little value unless the analytical sample is representative of the bulk from which it was taken. The magnitude of this problem may be realized by considering that the characteristics of many kilograms of material are assumed based on analyses carried out on the gram or even milligram scale (Figure 1).
There are issues and challenges involved in the sampling and characterization of pharmaceutical mixtures. Accurate and reliable characterization is hindered by both the complexity of granular systems and the lack of validated and reliable sampling technology and techniques1.
A discussion of drug sampling is beyond the scope of this article; the reader is referred to a recent review paper on this subject2.
 Sampling Procedures
Sampling Procedures
For most pharmaceutical materials (especially powders), several factors must be considered when devising a sampling scheme. These factors include the nature of the ensemble from where the sample is to be drawn, the cost of the sampling and associated assay(s), convenience, and the degree of precision required in the final result. The following issues need to be addressed when developing, or adapting, a sampling procedure: (i) the quantity of powder from which the samples are to be obtained, (ii) the amount of sample required, (iii) the powder characteristics including flow behavior, particle shape, tendency to segregate and surface chemistry, and (iv) mechanical strength (friability).
Statistical Sampling
Sampling procedures can be supplemented using established and proven mathematical techniques3. The advantage is that inspection of only a small fraction of the bulk is needed, which greatly reduces the amount of time and effort in sampling. Further, the level of certainty in detecting mistakes upon applying a given level of inspection can be calculated4. It is obvious that the larger the sample size, the smaller any errors will be. However, the size of the laboratory sample is typically minute compared to the bulk material and is subject to a large degree of variation. This can be reduced by creating a stock sample from many increments from the bulk material (a minimum of 20 has been suggested) and then dividing it down. The larger the PSD, the greater is the potential for significant sampling errors.
Segregation
Segregation is the biggest problem affecting sampling of powders and gets increasingly worse as the material PSD broadens. The degree of segregation is also affected by the flow characteristics of powders and is significantly worse for free-flowing materials.
During storage of materials, fine particles move, due to vibrational energy, into the interstitial spaces between large, coarse particles, forcing them to rise over time to the top of the container. Samples should never be taken from the surface region of any stored material. When powders are poured out of any container onto a surface, the inrush of air into the container causes any surface fine particles to be expelled into the atmosphere away from the bulk of the material. Then, as the powder begins to “heap” on the surface, it starts to “unmix” since the fine particles tend to stay closer to the center of the heap while the coarser particles segregate to the peripheral regions of the heap. Thoroughly mixing of any stored material and then sampling from the poured stream of material can minimize bias.
The propensity of a powder to unmix also affects drug product homogeneity5, for example, a combination of a micronized API with much coarser filler/excipient. To minimize this problem, the difference in PSD of the two materials should be less than 40%.
Cohesive powders, sticky or moist material, and fibrous solids do not segregate readily. Passing these materials through a mixer or sieve before sampling is recommended.
Sampling methods
Static sampling
There are three basic methods: (i) scooping, (ii) thieving, and (iii) cone & quartering. Scoop sampling is widely used and consists of plunging a scoop into a heaped batch. This method is prone to error because the whole sample does not pass through the sampling device. As mentioned previously, since the sample is taken from the surface it may not be representative of the bulk. Thieving consists of plunging a capture device (comprising one or more separate sample chambers) into the bulk material to retrieve several small aliquots of the powder. The sample chamber(s) can be opened and closed by an operator via controls at the top of the device. However, because large particles will flow more easily than small particles, an opened thief is liable to be filled preferentially with the coarse fraction of the PSD. Also, there can be problems with damage to friable or fragile (i.e., needle-like crystalline) materials. While thieving is a significant improvement over scoop sampling, it is only an adequate method and is considered an inferior technique6,7. Cone & quartering requires that the powder be poured as a cone-shaped heap onto a flat surface. The heap is flattened and divided by a cross-shaped metal cutter. One fraction (¼) is taken and the whole procedure is repeated until the specimen size desired is obtained. This technique is strongly operator-dependent and is not recommended.
Dynamic sampling
Superior methods of powder sampling are obtained by using procedures where the sample is removed from a moving powder bulk. Again, there are three basic methods: (i) table sampling, (ii) chute splitting, and (iii) spin riffling. Table sampling involves powder flow along an inclined table in which there are a series of holes. Guides, in the shape of prisms, placed in the path of the powder stream break it into fractions. Some powder falls through the holes and is discarded while the powder remaining on the incline plane passes onto the next row of prisms and holes and more is removed. This process is continued and the powder that is collected at the end of the table is the sample. Unfortunately, error results because the initial feed is not uniformly distributed and complete mixing after each separation cannot be achieved. The chute splitter consists of a V-shaped trough along the bottom of which is a series of chutes alternately feeding two trays placed on either side of the trough. The sample is poured into the chute and repeatedly halved until a sample of desired size is obtained. Increasing the number of chutes in the device may increase the efficiency of the system, but there are limitations. However, when the system is appropriately designed and validated, its use can yield satisfactory sample division. A spinning riffler device uses mechanical (vibrational) energy to provide a constant flow of material from its holder. The steady flow passes through a divider head that rotates at a constant speed, thus minimizing segregation. The amplitude of vibratory motion and velocity of circular motion can be controlled separately. This allows different powders having varying flow characteristics to be subdivided. Each sample from this type of device can be analyzed separately or recombined to yield a representative sample.
Comparison of Sampling Techniques
Table 1 summarizes the advantages and disadvantages of the techniques. The relative efficiencies of five common sampling procedures are summarized in Table 28. In the study, reproducibility of the technique was based on the standard deviation in size distribution observed between different subsamples generated from the same primary sample. The spin riffler method is clearly superior to all other methods and this has been confirmed by several other studies9 -11.
 Conclusion
Conclusion
Since powder batches can vary from a few grams to many kilograms, there are various devices and techniques that have been developed to aid in representative sampling. Wherever possible the two Golden Rules of Sampling should be followed (Table 3) 12.
Together with statistical sampling procedures these can provide a high degree of reliability and homogeneity in the samples produced. Some techniques are fairly independent of the operator while others are strongly influenced by human factors. Whatever technique used, one should develop and implement practices and controls in sampling procedures that document and track any error or variation that may be introduced by human factors. If possible, the spinning riffler method should be used.
References
- J. Muzzio et al, Int. J. Pharmaceutics, 250 51 (2003).
- USAID, Guidelines for Drug Sampling, USP DQI Drug Quality Monitoring Program, August (2006).
- Herdan, Small Particle Statistics, Butterworths, London (1960).
- ISO 2859, Statistical Sampling, International Organization for Standardization, Geneva (2000).
- Train, Pharm J., 185, 129 (1960).
- Berman, A. Schoeneman, and J.T. Shelton, Ind. Pharm., 22 1121 (1996).
- P. Garcia, M.K. Taylor, and G.S. Pande, Pharm. Dev. Tech., 3 7 (1998).
- Allen and A.A. Khan, Chem. Engr., 238 CE108 (1970)
- R. Montgomery, Analyt. Chem., 40 1399 (1968).
- H. Kaye, Powder Metall., 9 213 (1968).
- A. Hatton, Powder Tech- nol., 19 227 (1978).
- Allen, Particle Size Measurement, 3rd Edition, Chapman and Hall, London, UK, (1981).