Quality by Design
In the past few years much has been said and written about Quality by Design (QbD)1,2. It has been discussed in many forums by the FDA and industry consultants as well. The FDA launched a pilot program in 2005 to allow firms an opportunity to submit chemistry, manufacturing, and controls (CMC) information obtained through the application of QbD3,4. However, only a handful of companies have submitted CMC information using the QbD model and it is still not well understood by industry. For the companies that chose to follow this path, there is an opportunity for a shortened review period.
Current Status
The International Conference on Harmonization’s Pharmaceutical Development Guidelines, ICH Q8, objective is to consistently deliver a high-quality product by building proper controls, understanding of your manufacturing process, and monitoring the right parameters and attributes1,2. It is a scientific, risk-based, holistic, and proactive approach to pharmaceutical development and is a deliberate design effort from product conception through commercialization with full understanding of how product attributes and processes relate to product performance.
Quality is not necessarily to be tested into the finished product, in fact the finished product may not be tested for all attributes. Under QbD principles, regulators will allow companies to rely on in-process and Process Analytical Technology (PAT) data to assure the manufactured product meets the established pre-determined quality attributes. Regulators’ expectations are that the information and conclusions related to the product, as well as the control strategy which assures that the product quality attributes are maintained, should be shared with them. The FDA’s Center for Drug Evaluation and Research has expressed their expectation regarding QbD as, “QbD is a systematic approach to product and process design and development”3,4. QbD involves the following key elements:
- Target the product profile
- Determine critical quality attributes (CQAs)
- Link raw material attributes and process parameters to CQAs and perform risk assessment
- Develop a design space
- Design and implement a control strategy
- Manage product life cycle, including continual improvement
Both industry and the FDA are continuing to focus on QbD as it relates to the acceptance criteria for real-time release. However, real-time release provides manufacturing flexibility and increased quality assurance and is a more modern approach to manufacturing and controls.
Design Space
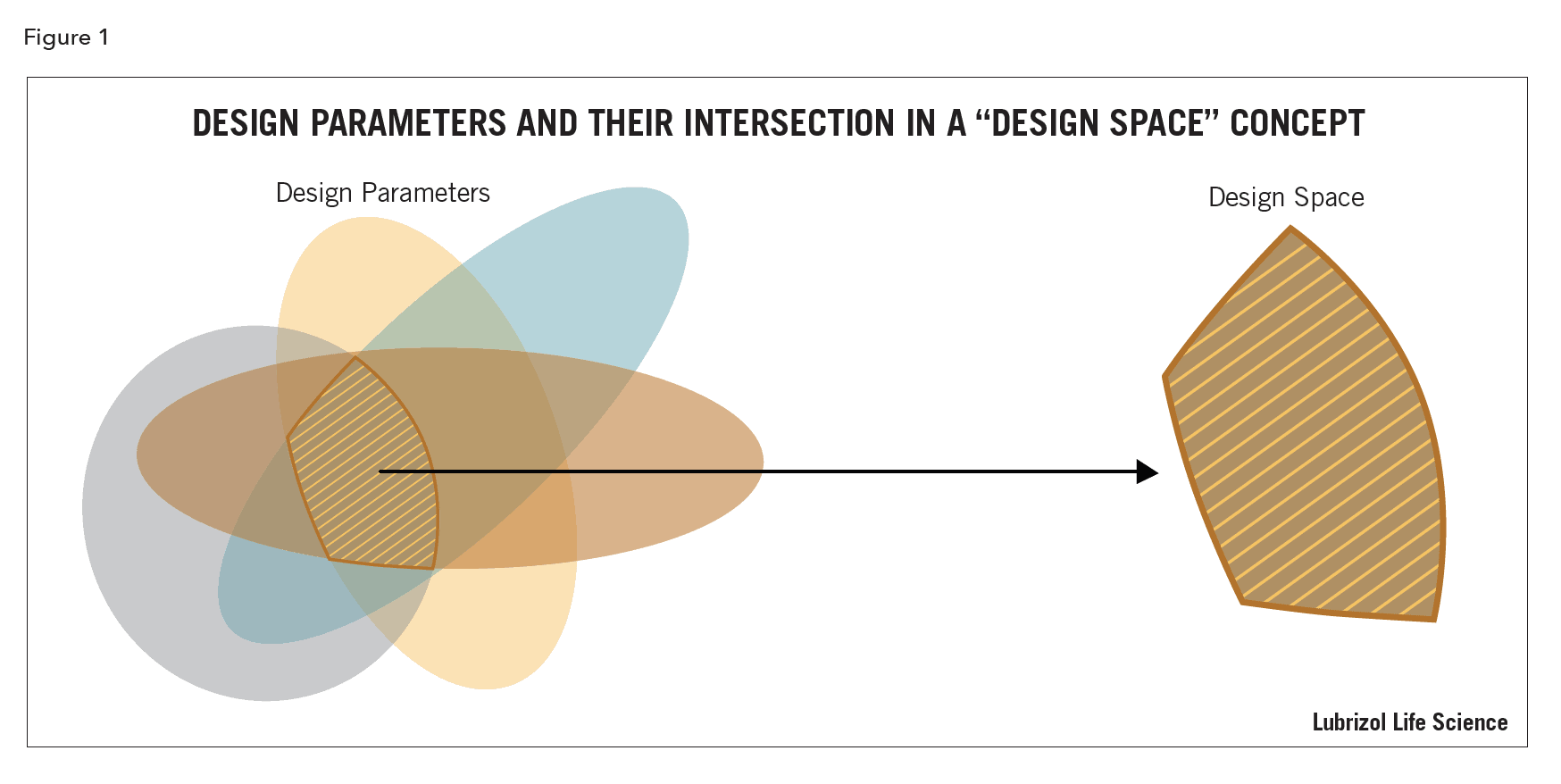
A systematic evaluation of the risks associated with manufacturing a pharmaceutical product is required for implementation of QbD. Expectations are that all raw materials and product variables with a potential impact on product quality are evaluated and assessed using adequate assessment tools, such as fishbone diagrams, Failure Mode Effects Analysis (FMEA), and Pareto analysis. Even though risk assessment is based on experience and process knowledge, when stating prior experience as a consideration in assigning or evaluating risks and interdependency among product manufacturing variables, adequate explanation should be provided as to how this prior experience is applicable3,4 to the product process control strategy3,4.
The process parameters with the highest impact on the CQAs should be further studied to better understand their impact on product quality. The control strategy that is implemented for risk reduction and control needs to consider these parameters and their impact on product quality for changes both within and outside the design space.
Variation in parameters is inherent to all processes and needs to be understood and controlled (Figure 1)5. The probability of falling outside of the design space per unit time needs to be evaluated so risk analysts can estimate probabilities of being outside (or inside!) of design limits, given various scenarios.
Evaluation of process parameters in a formal risk assessment and implementation of control strategies to define a design space helps increase assurance of product quality by reducing variability.
The FDA has indicated risk assessment is a powerful tool and is important for effective communication between the FDA and industry and for intra-company communication (such as between R&D and manufacturing or among multiple manufacturing sites)3,4.
The design space should be determined for raw materials as well as for the finished product so that interactions are well understood, and variability controlled. This ensures CQAs are not adversely affected and expected drug product quality is reproducible.
What does all that information provide?
- It demonstrates knowledge of the product
- It identifies possible sources of variability and how associated risks can be mitigated
- It allows for the assessment of the product quality attributes when one or more of the variables fall outside of the design space
- It forms the basis for continuous improvement
 Control Strategy
Control Strategy
Control strategy, including Continuous Quality Overall Summary (CQOS), is also a critical element of QbD, and should include starting materials, intermediates, and finished products. The strategy should include every aspect known to potentially impact the product. The following are suggested considerations for a control strategy:
Manufacturing instructions:
- Set the most appropriate parameters
- Multivariate interactions need to be evaluated
- Correct ranges of operation
- Determine the proper data for trending and analysis
Create a control strategy:
- Control the quality of product & components
- Scale-up and equipment impact
- Technology transfer considerations
- Control critical processing parameters
- Implement PAT strategies, if possible
- Trend and analyze the right data
- Implement adequate supply chain practices
The collection of this data helps in controlling variability and minimizing risk as well as providing a CQOS which will serve as the basis for continuous improvement during the product lifecycle.
Drug developers will need to pay attention to and consider these requirements during development. This will include more stringent application of design of experiments (DOE), focusing on systematic thinking, and organizing data in to meaningful categories. All of this leads to positioning the decision-making process to be based on the data and knowledge obtained from all stages of development.
Product Lifecycle
During the product life cycle, strong quality systems, mentioned below, are certainly critical in maintaining adequate controls and feedback to assure product quality.
Quality systems:
- Change control
- Validation
- Trending and analysis
- Management review
- Quality risk management
- Operator training
- Product improvement program
The implementation of Quality Systems in the manufacture of drug products helps pharmaceutical companies:
- Improve quality of pharmaceutical products
- Improve cGMP compliance
- Facilitate continuous improvement
- Implement & effectively utilize:
- Quality by Design (Q8 Pharmaceutical Development)2
- Risk Management (Q9 Pharmaceutical Risk Management)6
- Effectively transfer knowledge
- CAPA
- Change control
- Trending and analysis
- Demonstrate a state of control
- Manage process parameters movement within the design space
- Identify adverse trends and implement adequate corrective actions
The information accumulated in the quest of fully understanding your product and its manufacturing process will allow you to predict the impact of potential variations in your supply chain during routine commercial manufacturing. This information may allow you to design better and more meaningful experiments when studying the impact of new materials and control systems for the manufacturing process.
ICH Q9 Quality risk management (QRM) requires that risk to product quality be evaluated using gained scientific knowledge of the system that produces the product6. The method of evaluation should take into account the level of risk, the severity of the event, the probability for occurrence of the event, and the detection systems in place providing an overall risk categorization. QRM will help improve product quality, GMP compliance, and facilitate continuous product improvement by focusing on those areas which have the greatest potential based upon their level of risk.
References
- ICH Q8(R1) Pharmaceutical Development, June 2009
- ICH Q8(R2) Pharmaceutical Development, November 2009
- Chi-wan Chen, “Implementation of Quality-by-Design Principles in CMC Review: Office of New Chemistry Approach”, Advisory Committee for Pharmaceutical Sciences, Oct 26, 2005
- Patricia Van Arnum, “A FDA Perspective on Quality by Design”, Pharmaceutical Technology Sourcing and Management, 3(12), Dec 5, 2007
- Gregg Claycamp, “ICH Q9: Quality Risk Management”, CDER Advisory Committee for Pharmaceutical Sciences, Oct 5-6, 2006
- ICH Q9 Quality Risk Management, Jun 2006