Development and Validation of In Vitro Release Testing for Semisolid Formulations
The measurement of drug release from a given dosage form is fundamental to drug product development. The specific technique employed is determined by the dosage form itself and the intended route of delivery. For solid dosage forms, dissolution testing has been used for the past 50 years1. More recently, for semisolid drug products, in vitro release testing (IVRT) has shown promise in evaluating release properties. An in vitro release rate can reflect the combined effect of several physical and chemical parameters, including solubility and particle size of the active pharmaceutical ingredient (API) and rheological properties of the dosage form2.
The most common IVRT method employs an open chamber design like the Franz diffusion cell system (Figures 1 and 2) and can be used with a synthetic membrane, a tissue construct, or biological sample, such as cadaver skin.
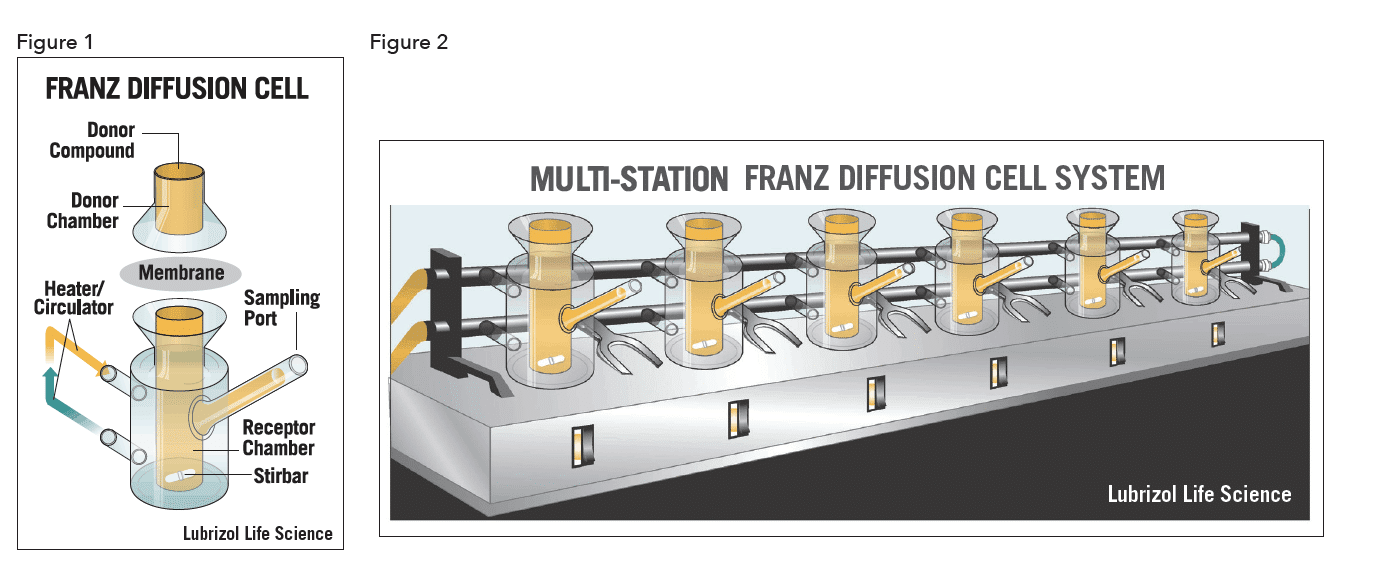
The membrane separates the donor compartment containing the test product from the receptor compartment filled with collection medium. Phosphate Buffered Saline (PBS) tends to be the collection medium of choice, though it may not always satisfy the requirements for a viable IVRT method. Diffusion of the drug from the semisolid product across the membrane is monitored by assay of sequentially collected samples of the receptor medium. At predetermined time points an aliquot of medium is removed from the receptor compartment for drug content analysis, usually by high-performance liquid chromatography (HPLC). The receptor compartment is topped off with fresh medium after each sampling.
In a previous article3, we discussed the challenges and issues related to IVRT for semisolid products. This technical brief summarizes developing and validating an IVRT method. A protocol for IVRT method development (usually accompanied by an appropriate analytical protocol) must be designed to satisfy both the developmental requirements and regulatory demands. Such protocols have been used to successfully validate IVRT methods4. The protocols test the method’s reproducibility, repeatability and robustness applying a rigid 90% confidence interval criterion. Illustrative IVRT parameters are given in Table 1.
 IVRT Method Development
IVRT Method Development
Successful IVRT is contingent on reliable drug transport from the test material through a membrane and into the receiving medium. Therefore, in identifying the optimal experimental parameters, the focus is on the API physicochemical properties and aimed at selection of the proper membrane, receiving medium, and sampling schedule, all being critical to a useful test.
For the receiving medium, the SUPACSS FDA guidance2 provides a reasonable starting point. The guidance states that an “Appropriate receptor medium such as aqueous buffer for water soluble drugs or a hydro-alcoholic medium for sparingly water-soluble drugs” can be used. Hence, after establishing a basic assay method, the first task in method development is to measure the solubility of the API in several solvents ranging from aqueous solutions such as PBS to hydro-alcoholic solutions such as isopropanol/ PBS-50/50 (v/v). The intention is to identify solvents that will provide sink conditions in the IVRT receiving vessel. Sink conditions exist when a receptor medium has a relatively “high capacity to dissolve or carry away the drug” and the receptor media “exceed[s] 10% of Cs (drug solubility in the releasing matrix) at the end of the test”5. Therefore, solvents yielding lower solubilities (<100 µg/ml) should be excluded from further IVRT development. Usually three media, including both aqueous-based and hydro-alcoholic-based solvents, are selected for further IVRT evaluation.
Since one purpose of IVRT is to, if possible, serve as a surrogate for in vivo testing, the medium most resembling the relevant physiological fluid is preferred, which is why PBS is often a first choice. However, for APls with low aqueous solubility, a modification of PBS is necessary to increase the solvency sufficiently to approach sink conditions. In addition to alcohols, surfactants, liposomal preparations, etc. may also be used to increase the solubility of the API in the receiving medium.
At times, two selections for the IVRT medium may be employed: one for QC purposes such as long-term stability tests and manufacturing process change control (in which case a non-physiologic medium may be used) and the other to mimic in vivo behavior using a physiologically relevant media. The latter medium may be useful for generating a potential in vitro – in vivo correlation.
There are many choices for membranes, which include recently excised tissue, tissue constructs, cadaver tissue, and synthetic membranes. Factors influencing the selection of the proper membrane include compatibility with the test material, availability, reproducibility, cost, and, importantly, the goal of the experiment itself. Synthetic membranes vary controllably in pore size, thickness, and hydrophilicity. Since the major constituent of many semisolid products is water, hydrophilic/hydrophilized synthetic membranes are typically used. During membrane screening, usually three polymeric membranes with the same pore size are evaluated. Combined with the release results from different media, an optimized experimental configuration is identified. More esoteric membranes may be of value at times. For example, if trying to determine the actual depth of penetration into skin for a particular API, cadaver skin may be used so that it could be subsequently dissected and analyzed.
Multiple sampling times (at least five) over an appropriate time period are recommended to generate an adequate release profile (i.e., at 30 min, 1, 2, 4, and 6 hours)2. Depending on the formulation’s burst effect and the time for release to reach a plateau, the sampling times may have to be adjusted. The “burst” is a common phenomenon where a high initial release is observed. It is also not uncommon to extend the run to 24 hours depending on the clinical use and desired experimental information.
The type of diffusion cell used will dictate other parameters such as the membrane diameter, test product application amount, and sample aliquot removed from the receptor chamber. As recommended by SUPAC-SS2, six cells are used to determine the release profile of each testing formulation/topical dermatological product. Like membranes, specialized cells can be employed to try to more closely mimic biologic systems. For instance, there are occlusive, flow-through, and multichambered configurations.
The data output of IVRT is a summary of release rates over a short time period (4 or 6 hours, whichever presents a straight line for the release figure, as required by SUPAC-SS2). Figure 3 shows a typical release profile.
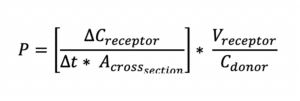
In addition to reporting release rates, other values such as the APl’s flux (µg/cm2 hr), 24-hour accumulation (µg/cm2), and permeability (cm/hr) can also be reported so that a more complete picture of API release and accumulation can be generated. Figure 4 shows the cumulative amount of API as a function of time.
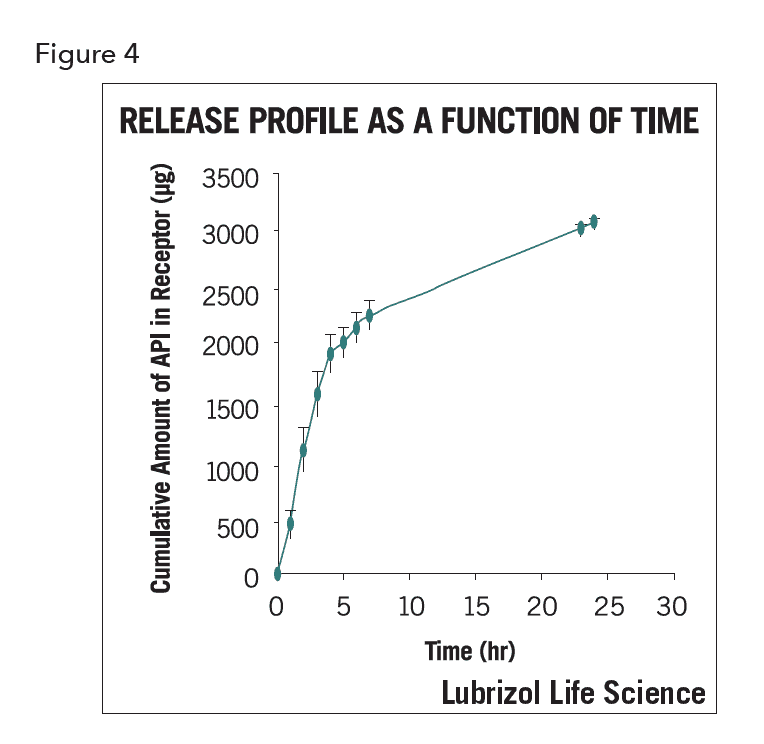
From this release profile the slope of the linear portion of early time release (i.e., the steady state) presents the flux of the API and the Y-axis labels the accumulation of API at certain times.
Permeability (P), defined as the rate of flow of liquid or gas through a porous material, is calculated by the following equation:
Validation is “a procedure to establish documented evidence that provides a high degree of assurance that a specific test will consistently produce a test outcome meeting its predetermined specifications and quality attributes”. A validated test is one that has been proven to do what it purports or is represented to do.2 In general, a specific statistical method is used to compare release results from different sets of validation runs. Such a method can be a simple standard deviation (SD) and relative standard deviation (RSD) comparison, or a Student’s t-test, among others.
The criterion used in SUPAC-SS, which is a 90% confidence interval (Cl) for the ratio of median release rates, is applied.2 The release results, i.e., results from the first 2 sets (12 cells, 2 runs of 6 cells) of validation runs are set as standard release rates5. For the rest of validation runs, a 90% confidence interval for the ratio of the median in vitro release rate (in the population) per validation run to the median in vitro standard release rate (in the population) should be computed, expressed in terms of percentage. If all 90% confidence intervals fall within the limits of 75% to 133.33%, the IVRT method meets the validation requirement.
During the validation phase, sets of experiments are performed under varied conditions to evaluate and demonstrate the reproducibility, repeatability, and robustness of the developed method. Those varied conditions include amount of gel applied, API strength, medium variation, temperature of water bath, etc.
Summary
IVRT provides an efficient method for the evaluation of drug release from semisolid drug products and with a properly validated method, to assess manufacturing quality over time. Its use is increasing as both a development tool and a means of setting product specifications. It seems likely that IVRT will soon become a required part of the regulatory package and, on a case-by-case basis, is already often included. Designing an appropriate IVRT system is critical for reliable information generation, to both help guide the development process and set useful drug product specifications.
REFERENCES
- Zhang, H, Yu, L, “Dissolution Testing for Solid Oral Drug Products: Theoretical Considerations”, Ameri can Pharmaceutical Review, September/October 2004, 7(5), 26-
- FDA Guidance for Industry: Nonsterile Semisolid Dosage Forms: Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Release Testing and In Vivo Bioequivalence Documentation, May 1997, SUPAC-SS CMC
- Fan Q, Mitchnick M, Loxley A, “The Issues and Challenges Involved in IVRT for Semi-solid Formulations”, Drug Delivery Technology, 2007: 7: 62-66. Full text: http ://wwwparticlesciences.com/docs/DDT_October_2007_150_DPl.pdf
- Particle Sciences Technical Brief Vol. 5: “Analytic Method Development and Validation
- The Topical/Transdermal Ad Hoc Advisory Panel for the USP Performance Tests of Topical and Transdermal Dosage Forms, Stimulus Article: Topical and Transdermal Drug Products, Pharmaceutical Forum, 2009: 35 (3):