
The Guide to Analytical Method Development
What is analytical testing and why is it essential for drug product development?
Analytical testing of a drug product describes the various techniques used to identify the physical and chemical characteristics of a given sample. The qualitative and quantitative results produced from analytical testing provides crucial information to control and confirm the properties of raw materials, active and inactive ingredients, and finished products.
In the early stages of a drug product development program, analytical testing provides information on how close initial prototypes are to the Target Product Profile (TPP). “The TPP is a document that provides a clear vision of the product objectives and guides R&D actions and decisions, including narrowing down drug candidates, designing clinical research strategies, and how to communicate with regulatory authorities such as the FDA,” notes Laurie.
As a project progresses, the analytical program evolves into ensuring the quality of clinical trial materials and, eventually, commercial drug products. Since many drug products are now outsourced, most of these materials are generated by contract development and manufacturing organizations (CDMOs). Substantiating the chemical and physical properties of an outsourced drug product requires a competent and hands-on partner.
Evaluation Criteria
Regardless of whether your product is developed in-house or with a partner organization, there are five crucial properties that analytical testing must evaluate and verify:
- Identity – was the proper product produced?
- Strength – is the right amount of active ingredient in each dosage unit?
- Quality – does the product meet the standard requirements?
- Purity – is the product free of contaminants and degradation products?
- Potency/Performance – does the drug behave as intended and have the desired effect?
Laurie draws on over twenty years of experience in analytical science when she says, “Knowing the answers to these questions is essential for assuring the safety and efficacy of a drug product. That’s why analytical testing is so important, and why knowledge of the individual tests and experience developing and validating analytical procedures is critical.”
What are the types of analytical methods?
There are numerous tests used to characterize drug products, but specific methods are selected based on individual project needs and TPPs. A knowledgeable formulator or development partner will work with you to create an analytical plan that outlines the different test types, when to perform them, and what information they will provide. According to Laurie, “Particle Sciences typically breaks analytical testing down into three categories—separation and quantitation methods, in vitro release methods, and physical characterization methods—and works from there to cultivate a unique strategy for each client.” The details of each category are as follows:
Separation & Quantitation Methods
Tests within this category include high-performance liquid chromatography (HPLC), gas chromatography (GC), mass spectrometry (MS), size-exclusion chromatography (SEC), and gel electrophoresis. These tests provide critical information on identity, strength, and purity, as well as the chemical stability of the drug over time.
In vitro Release Methods
Dissolution, in vitro elution, and in vitro release testing comprise the primary methods utilized within this classification. These tests present critical information on the consistency of drug release from the drug product matrix. They also aid in predicting relative drug availability and in vivo delivery of the active pharmaceutical ingredient (API).
Physical Characterization Methods
Physical characterization tests include those for particle size/charge, molecular spectroscopy, X-ray powder diffraction (XRPD), and differential scanning calorimetry (DSC). These tests are vital as they provide information on physical attributes like solid-state form, viscosity and elasticity, particle size, mechanical properties (e.g., tensile or compressive forces), stability, and aesthetics of the drug product.
Figure 1: Types of Analytical Methods Developed at Particle Sciences

How does stage in the development process impact analytical methods?
One of the many factors an experienced partner will account for when creating an analytical strategy is the current and upcoming phase(s) of product development. Devoting time early on to cultivating sound methods establishes a strong foundation that facilitates validation when moving to larger-scale production down the road, ultimately saving a developer time and money.
Proof-of-Concept / R&D
This initial stage in development is where a formulator first lays out analytical procedures to support the TPP. The tests performed during this phase aid in understanding the underlying behavior of the API on its own and as it interacts with other formulation components (e.g., excipients, polymeric delivery systems, stabilizers). They also provide feedback to the formulator regarding how closely the prototype matches the TPP.
Early Development
The second phase is where an analytical chemist obtains initial information on process/product repeatability and robustness. Analytical testing performed here assists a developer in understanding the short-term stability of the API and the overall drug product under both normal and stress conditions. Analytical testing during early development also aids in identifying the critical quality attributes (CQA) of the drug product and obtaining rank order drug release rates from prototypes, which are crucial pieces of information for optimizing drug product performance.
Engineering Batch and Clinical Trial Manufacturing
Since the CQAs will have been set before this stage, analytical testing on clinical trial material is performed to measure the CQAs. At this point, the CQA criteria will be relatively broad, but recording the attributes with the appropriate analytical methods to ensure they are within the desired specification range is still vital. These analytical methods should include, for example, collecting long-term real-time and accelerated stability data to assign shelf-life. An informed formulator or CDMO will also assess the reproducibility of the manufacturing processes at this stage.
Commercial Manufacturing
The specification range for CQAs is significantly narrowed when a product reaches commercial manufacturing due to the work performed in the previous stages of product development. The role analytical testing plays during commercial production is primarily to monitor the CQAs and long-term stability as well as to assess the effects of any process, material, or supplier changes on the CQAs.
“Each phase in a product’s lifecycle has unique analytical considerations, and the respective testing conducted provides vital and diverse information for drug product development,” states Laurie. “A reliable and experienced partner understands how to design a phase-appropriate method validation strategy, which helps enable efficiency and cost reduction.”
Figure 2: Phases of Analytical Services at Particle Sciences

How does the Particle Sciences approach analytical testing?
Particle Sciences specializes in complex formulation challenges and often takes on projects requiring specialized knowledge or techniques during development and manufacturing. To overcome these development hurdles, proper characterization and monitoring of product properties are essential.
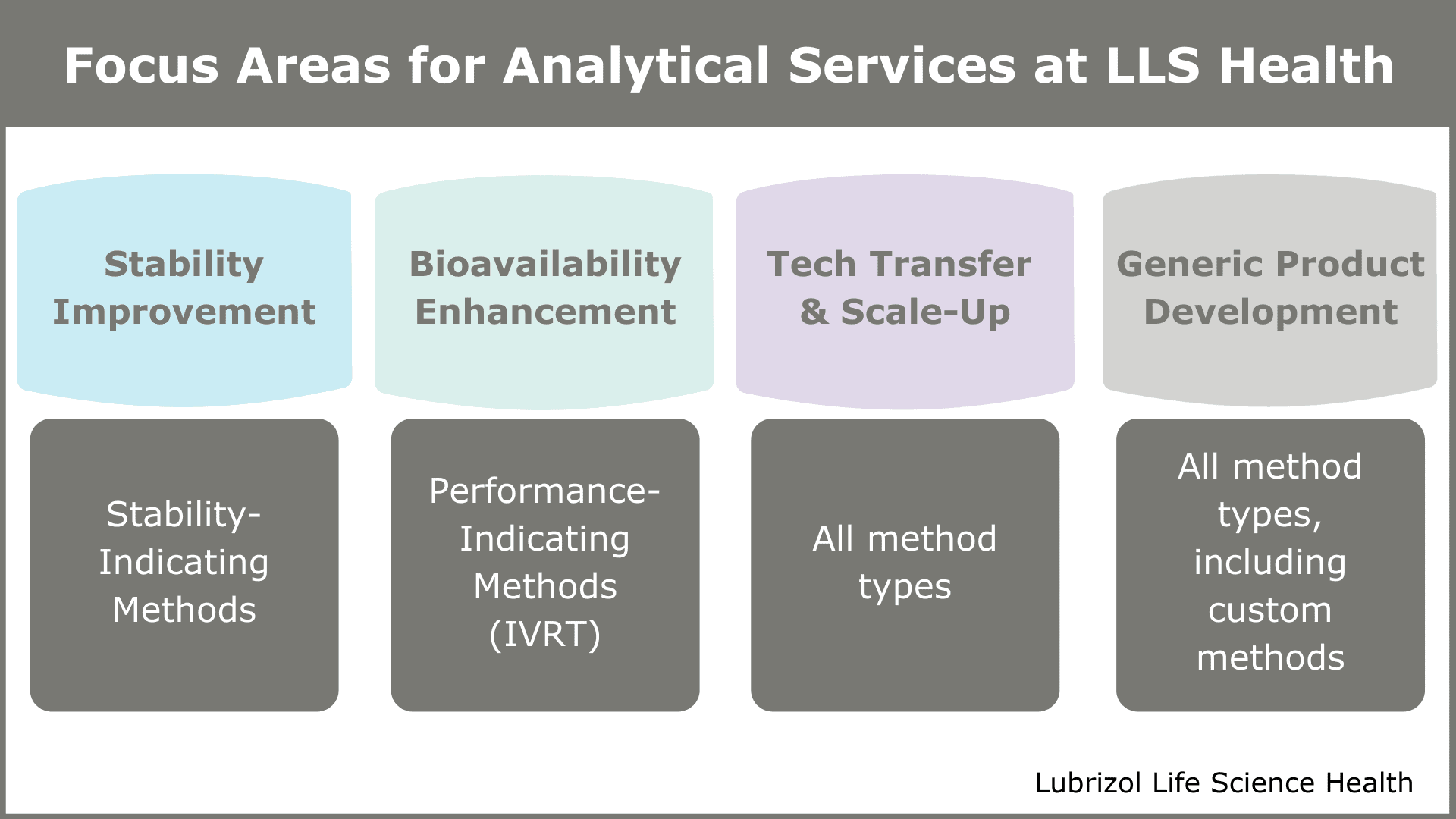
“The projects we encounter typically are characterized by at least one of four elements – they either seek to improve stability of an API, increase the bioavailability of an API, replicate an existing drug product (i.e., formulate a generic), or perform a tech transfer production scale-up,” says Laurie. “Based on the unique challenges presented with each of these, there are certain analytical methods that may take on increased importance.”
Stability Improvement Projects
Stability is a common issue with drug products and formulators often look to CDMOs such as Particle Sciences to help identify solutions. For this to be possible, accurately measuring stability and monitoring it throughout the product lifecycle is essential, meaning stability-indicating methods are key for this type of project. These methods distinguish critical changes in chemical or physical characteristics, such as the presence of API degradation products, which is necessary for determining the overall acceptability of a product.
Bioavailability Enhancement Projects
Discriminatory in vitro release tests are the primary tool utilized with bioavailability enhancement projects and are used to rank order prototypes in the proof-of-concept stage. Later, these in vitro release tests may be optimized to distinguish the effect of a critical quality attribute on target release rate—for example, viscosity in a topical formulation, sheath thickness for a core-sheath device with a rate-controlling membrane, or encapsulation efficiency for a solid-lipid nanoparticle. Most APIs in the drug product pipeline today are BCS Class II or IV compounds that require bioavailability improvement or alteration of their pharmacokinetic profiles. Formulation approaches such as nanomilling may improve the solubility of a poorly water-soluble API, leading to increased bioavailability. However, improving the solubility of a poorly water-soluble compound while maintaining a stable, narrow particle size distribution requires expertise in both formulation and analytical characterization of particulate suspensions.
Technology Transfer or Scale-Up Projects
When scaling-up or tech transferring a manufacturing process, demonstrating that the CQAs remain consistent with the reference product is essential. Analytical methods, via chemical, physical, and release testing, provide this information. When designed and executed correctly, analytical testing helps facilitate a smooth transition. At Particle Sciences, we are uniquely positioned to transfer in proprietary or academic technologies/processes and bring them into GMP production, and our analytical services team plays a major role.
Generic Development Projects
In generic product development, formulators utilize a full suite of analytical methods to assure sameness to the predicate product, which the FDA defines as Q1/Q2 sameness and Q3 similarity. The FDA also provides numerous guidance documents with further instruction on conducting in vivo bioequivalence (BE) studies for generic drugs, and a growing number for in vitro BE. In instances where generic drugs must undergo in vivo testing, a well-designed in vitro test informs the in vivo study, which can save considerable amounts of time and money in the approval process.
“So, taking into consideration the product lifecycle stage along with the various types of analytical testing and the information they provide, we factor in the project type to form an analytical approach that best supports client goals,” says Laurie, “Analytical testing really is vital to the development success of a drug product, and we continually witness this firsthand with our partners.”
An Overview of Q1, Q2, and Q3 Sameness and Similarity Requirements
For generic drug products delivered via the parenteral, ophthalmic, and otic routes, the FDA requires Q1 and Q2 sameness for generics. Q1/Q2 sameness may also be recommended in a product specific guidance (PSG) when in vitro methods for demonstrating bioequivalence are proposed. The definitions of Q1 and Q2 sameness are as follows:
- Q1 (Qualitative sameness): the test product uses the same inactive ingredient(s) as the reference listed drug (RLD)
- Q2 (Quantitative sameness): concentrations of the inactive ingredient(s) used in the test product are within +/-5% of those used in the RLD
Q1/Q2 sameness may be demonstrated with a formulation table showing the inactive ingredients and their quantities relative to an RLD. However, for some complex ANDAs, Q3 similarity must also be considered:
- Q3 (Physicochemical similarity): the arrangement of matter (physical and structural properties) is similar to the RLD
Q3 similarity mitigates the risk associated with physicochemical variations in a generic drug product, including, but not limited to:
- Differences in the polymorphic form of a drug
- Differences in rheology
- Differences in crystallinity that can impact drug diffusion
When evaluating products such as ophthalmic suspensions and multi-phase creams, these physicochemical properties can have a significant impact on the performance of the generic drug product. As a result, many PSGs, especially those for topical ANDAs, recommend Q3 evaluations.
At Particle Sciences, our fully-equipped physical characterization lab provides measurements of particle size distribution (laser diffraction and dynamic light scattering), rheological analysis, and solid state characterization. We also have in-house expertise with HPLC, FTIR, DSC, TGA, LC/MS, Raman microscopy, and other methods that support evaluations of key properties such as crystallinity and solubility. Our breadth of unique analytical capabilities positions us as an ideal partner for complex generic drug product development and ensure we can help you meet Q1, Q2, and Q3 requirements.
Figure 3: Focus Areas for Analytical Services at Particle Sciences
Future trends and outlook of analytical method development
Products in the pharmaceutical pipeline have become increasingly complex—a trend that will likely continue. API bioavailability and stability issues are on the rise, as well as the development of intricate generic, biologic, and aseptically manufactured drug products. With this uptick in complexity, identifying and measuring the key physical and chemical characteristics of a product is an increased challenge.
When choosing whether to bring in a CDMO, it’s important to remember that identifying a comprehensive partner is about more than just possessing the right equipment – decades of know-how, knowledgeable staff, and demonstrated success is key.
“With so many formulation challenges on the development horizon, choosing an outsourcing partner such as Particle Sciences with so much experience overcoming these hurdles and accelerating our client’s products to market really can make or break a project” concludes Laurie.
Contact us today to see how our experienced analytical team can make a difference in your product development program.
Authors: Laurie Goldman and Ashley Rein