Key Takeaways From the FDA Complex Generic Drug Product Development Workshop: Part 1
Key Points
- The FDA is taking steps to simplify complex generic drug product development and increase competition
- Through the Pre-ANDA Meeting Program and new product-specific guidances, the FDA is clarifying scientific and regulatory requirements for ANDA filings
- The Agency is using learnings from nearly two years of product development meetings to guide potential applicants
The FDA’s Focus on Complex Generic Drug Products
On September 25-26, 2019, the FDA hosted its second annual Complex Generic Drug Product Development Workshop. As a continuation of the 2018 workshop, the two-day event consisted of presentations covering:
- GDUFA research on complex products for product-specific guidance development
- Pre-ANDA meetings and review process
- Various areas of complex product science, including development and characterization considerations
Complex drug products are critically important from a therapeutic perspective, as they enable the treatment of many chronic conditions and overcome challenges such as poor patient compliance. As an example, the table below shows the 29 long-acting injectable products that have received FDA approval. These products treat several serious conditions, including serious central nervous system disorders and cancers. Additionally, many of the products on this list are blockbuster drugs with over $1 billion in annual sales. However, only one of the products below—Depo Provera®—has an FDA-approved generic!
Table 1: FDA-Approved Long-Acting Injectable Drug Products
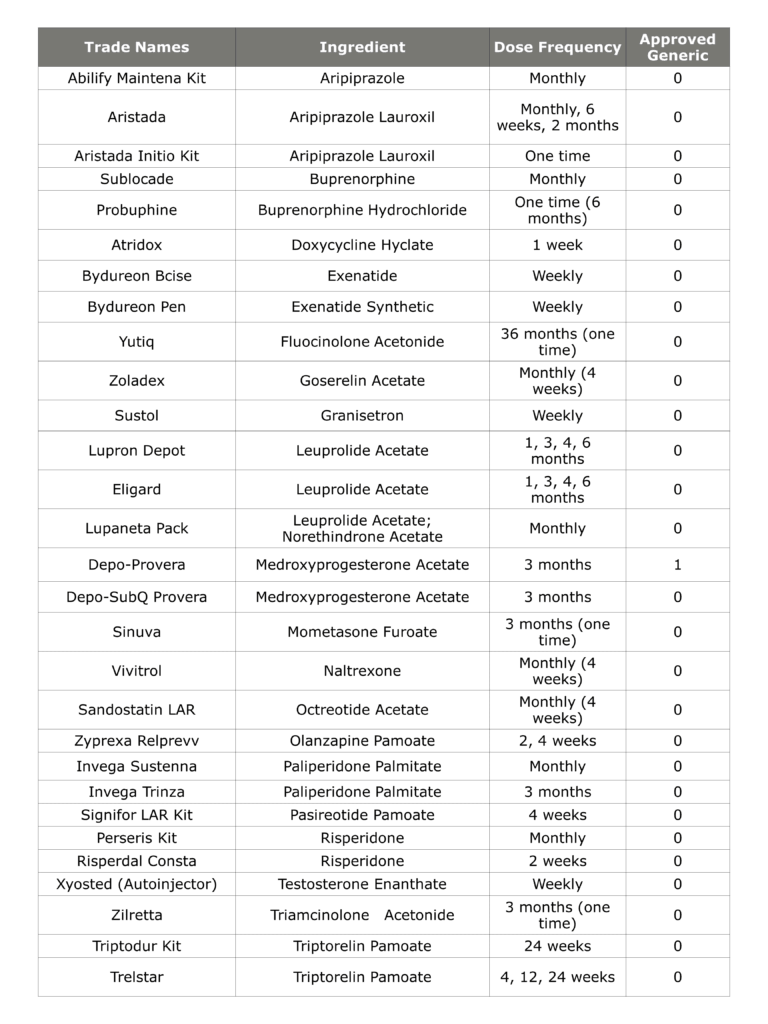
As this example shows, there is both a clinical need and a commercial motivation to develop generic versions of complex drug products such as long-acting injectables. Despite this, complex products have little to no generic competition due to the scientific, legal, and regulatory challenges associated with their generic ANDAs. Developing any generic medication requires a significant investment of both time and resources, but the elevated risks of a complex generic can deter even the most innovative companies.
As a result, the FDA has put increased focus on bringing complex generic drug products to market. In addition to several policy initiatives in recent years, the FDA has organized workshops and town halls to communicate how research outcomes guide and facilitate complex generic drug product development. The strategy has worked well to date, as the Agency approved a record number of generic drug applications in 2018 and 2019.
In this two-part series, we will review takeaways from the 2019 FDA Complex Generic Drug Product Development Workshop and convey how the FDA is working to streamline complex generic approvals and increase the number of ANDAs on the market.
First, What is a Complex Generic Drug Product?
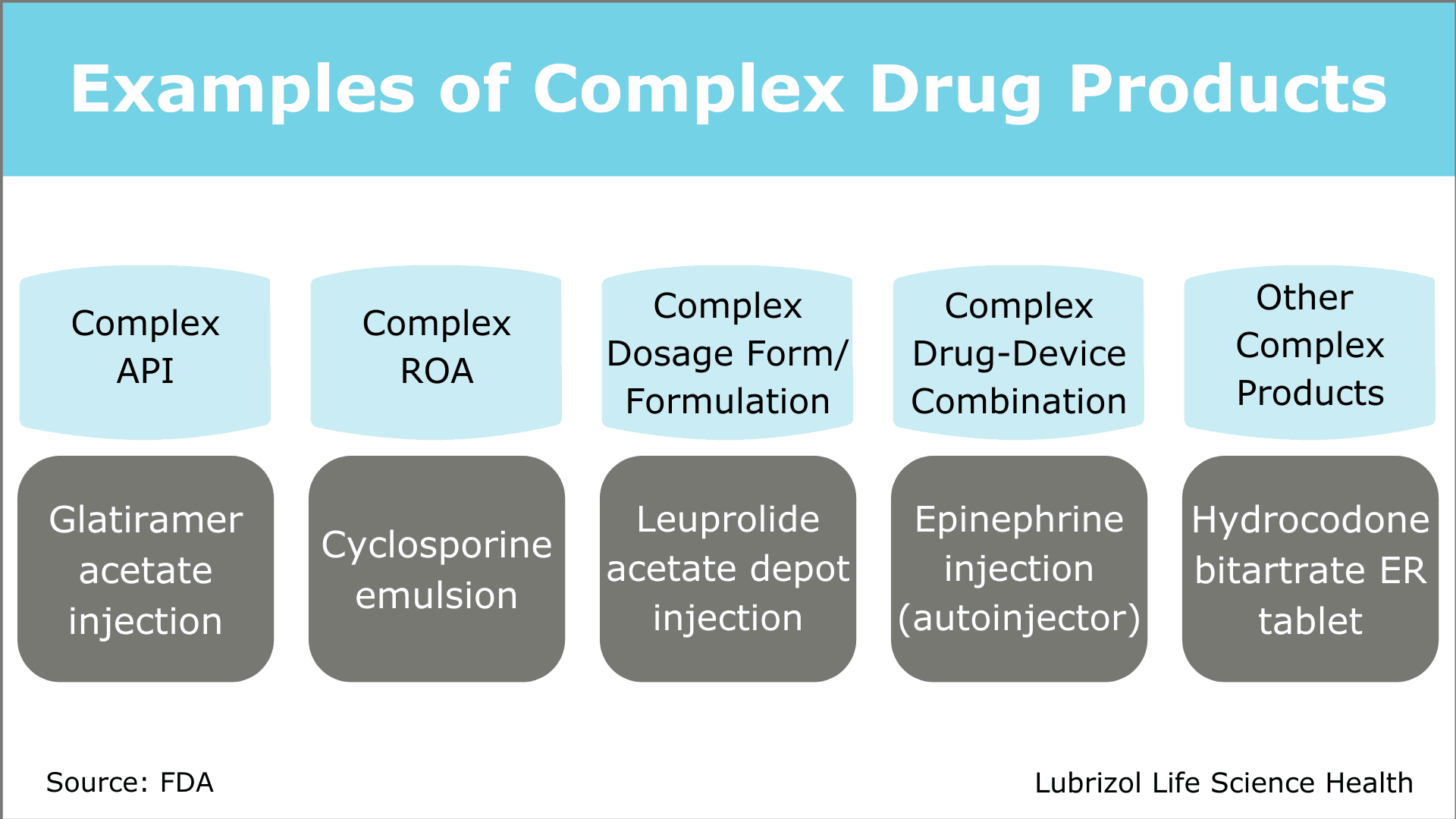
According to the FDA’s 2016 GDUFA II Commitment Letter, a complex generic drug product is generally defined as one or more of the following:
- A product with:
- a complex active ingredient(s) (e.g., peptides, polymeric compounds, complex mixtures of APIs, naturally sourced ingredients)
- a complex formulation (e.g., liposomes, colloids)
- a complex route of delivery (e.g., locally acting drugs such as dermatological products and complex ophthalmological products and otic dosage forms that are formulated as suspensions, emulsions or gels)
- a complex dosage form (e.g., transdermals, metered-dose inhalers, extended-release injectables)
- Complex drug-device combination products (e.g., auto-injectors, metered dose inhalers)
- Other products where complexity or uncertainty concerning the approval pathway or possible alternative approaches would benefit from early scientific engagement.
Complex generic drug products span a range of dosage forms, routes of administration, and API types (Figure 1). Challenges associated with developing complex drugs can be highly specific or generally applicable to many products. In the following sections, we will explore how the FDA has worked to encourage complex generic development and summarize recommendations for successful development programs.
Figure 1: Examples of Complex Drug Products
Product-Specific Guidances Continue to Grow
The Generic Drug User Fee Act (GDUFA) was introduced in 2012 to create an evidence-, research-, and science-based standards setting program for the FDA. In the first few years of GDUFA, approximately 800 product-specific guidances (PSGs) were published. These documents “identify the methodology for developing generic drugs and generating evidence needed to support generic approval.” PSGs contain recommendations on complex in vitro and in vivo release testing, among other topics. The program proved successful, leading to more generic drug approvals and higher success rates of generic drug applications. As a result, the program was revised in 2017 (GDUFA II) to expand the scope of FDA communication with industry.
In GDUFA II, the FDA defined complex drug products, created an expedited review process for drugs with limited competition, and established a pre-ANDA program to:
- Assist applicants to develop more complete submissions
- Promote a more efficient and effective ANDA review process
- Reduce the number of review cycles
- Facilitate approval of complex generic products
The pre-ANDA program included a renewed commitment to publishing PSGs. As of July 2019, approximately 1,700 PSGs were available, and in 2019 alone, over 160 new and revised PSGs were published (Figure 2). The introduction of PSGs has streamlined ANDA development, leading to more complete generic drug applications and a reduction in the average number of review cycles required to obtain approval. The FDA continues to publish new and revised PSGs on a quarterly and as-needed basis, incorporating both Agency research and industry comments to educate complex generic developers.
Figure 2: FDA Product-Specific Guidance Statistics
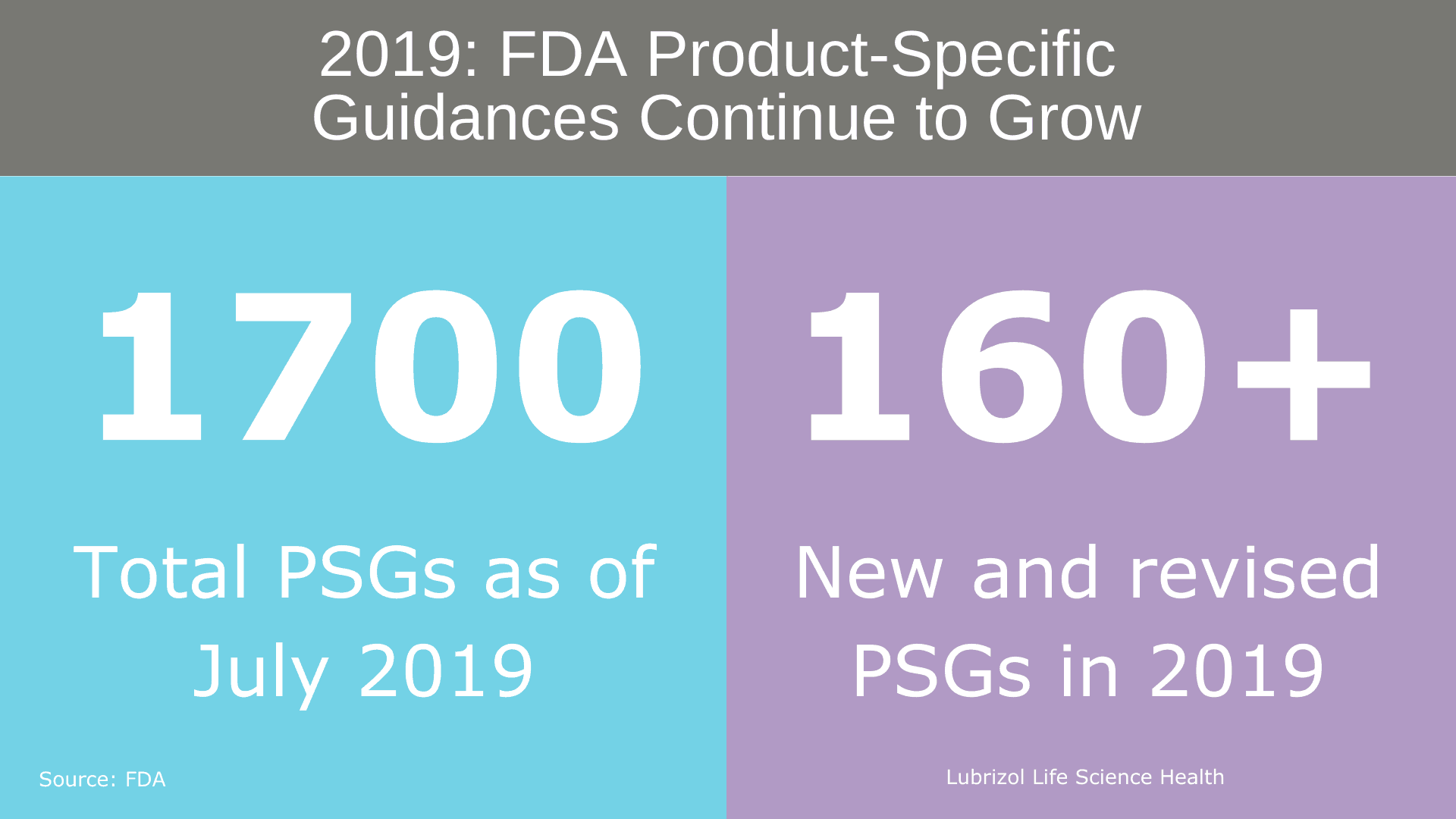
Pre-ANDA Meetings are Welcomed and Encouraged
In addition to PSGs, GDUFA II created a pre-ANDA meeting program to enable direct feedback from the FDA on ANDA submissions. These meetings allow applicants to ask questions that aren’t addressed in a PSG, propose alternative bioequivalence study designs, and receive Agency input on complex development issues. There are three types of meetings that may occur:
- Product Development Meetings: Provide for discussion of specific scientific issues or questions, such as an alternative bioequivalence approach. Give applicants the opportunity to receive targeted advice from the FDA on their development plan and early-stage data
- Pre-Submission Meetings: Provide an opportunity for prospective ANDA applicants to discuss and explain the format and content of the ANDA to be submitted. Allow the FDA to identify items or information that need further clarification before the submission takes place
- Mid-Review Cycle Meetings: Afford an opportunity for the FDA to discuss issues identified during review with the applicant
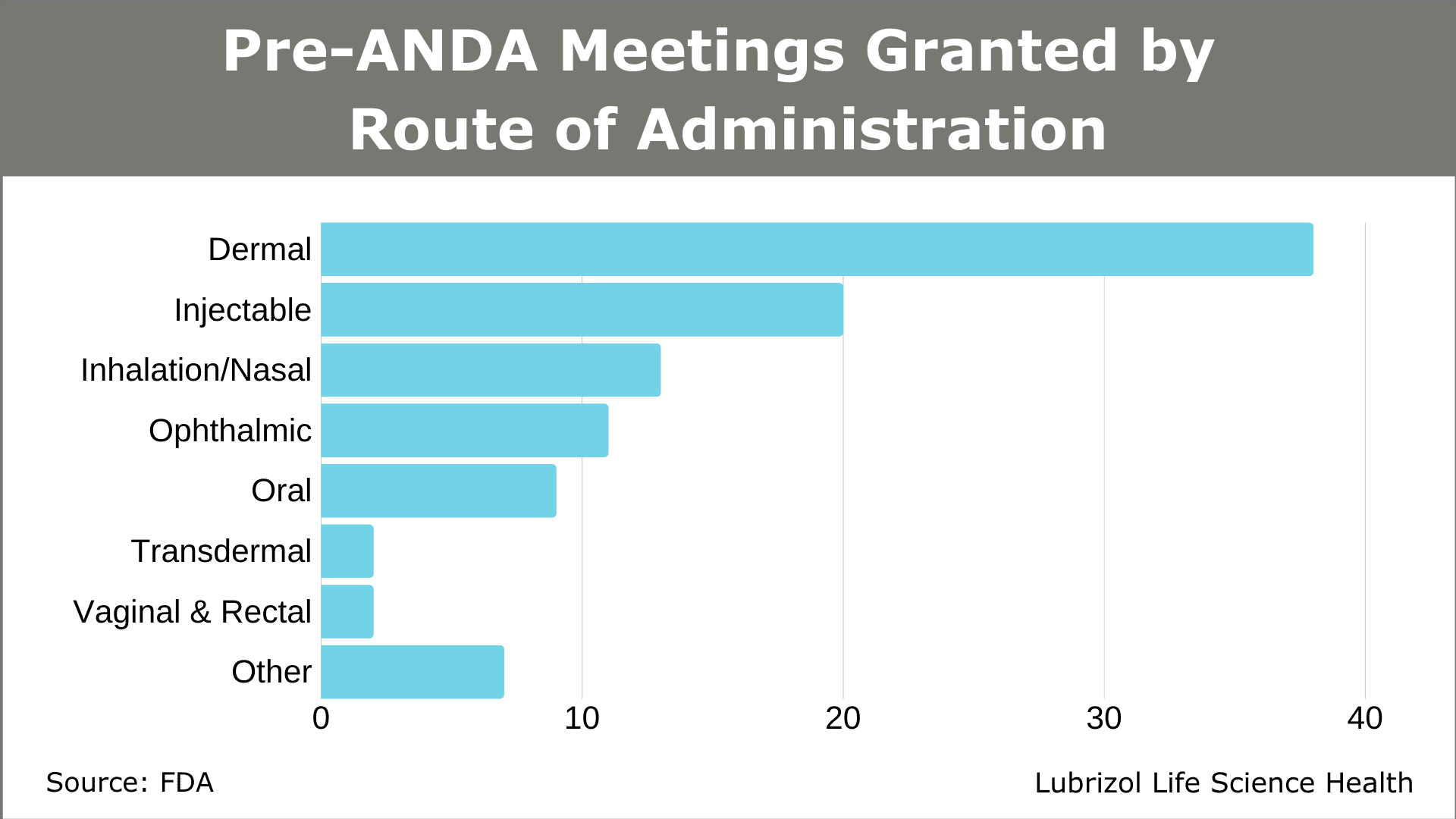
A growing number of drug developers are taking advantage of this resource. In 2018, the first full year of the pre-ANDA meeting program, 50 meetings were granted. The total was surpassed in just Q1 and Q2 of 2019 (53 meetings granted). Meetings have covered several routes of administration, with dermal, injectable, inhalation/nasal, and ophthalmic products accounting for most of the submissions through June 30, 2019 (Figure 3).
Figure 3: Pre-ANDA Meetings Granted by Route of Administration
With nearly two years of meetings to draw upon, the FDA also presented several examples of effective pre-ANDA questions to guide potential applicants. These included:
- Are there additional critical material attributes or critical process parameters that FDA feels we should address?
- Does the Agency agree that, on the basis of the data presented, the proposed physicochemical tests are appropriate to support comparative physicochemical characterization?
- Does FDA concur with proposed alternative in vitro approach to demonstrate bioequivalence?
- Are there any additional in vitro characterization studies recommended by the Agency?
Conclusion and Looking Ahead
Together, PSGs and pre-ANDA meetings create a comprehensive program for obtaining guidance on the scientific issues that accompany complex generic development. Developing complex generic drug products is not trivial, but the FDA’s continued commitment to these routes of communication has clarified the Agency’s expectations, ensuring developers generate the scientific data needed for approval.
If you are looking for a partner to develop a complex generic drug product of your own, look no further than the experts at Particle Sciences. We have decades of experience across a range of dosage forms, including microparticle and gel depot injections, implantable systems and combination products, and many sterile ophthalmic products. Have a project in mind? Contact us today.
In part two of this post, we address four additional takeaways that demonstrate how the FDA is actively investigating complex scientific topics related to complex generic drug products, including API/excipient characterization, particle size analysis, and in vitro drug release testing.
Click here to read part two of this post
Authors: Joey Glassco